The Bioinformatics Research Laboratory (BRL), develops new experimental and computational methods for discovery through genomics, epigenomics, and informatics. BRL aims to enable new discovery methodologies employing massively parallel sequencing and other high-throughput genomic technologies.
A recent research initiative is the construction of the Epigenome Atlas as part of the NIH Epigenomics Roadmap Initiative. The BRL along with the Epigenome Center serves as the designated Epigenomics Data Analysis and Coordination Center (EDACC) for the Initiative. The broad aim is to understand the variation of epigenomes (methylation status, histone marks, chromatin accessibility) across major human tissue types and experimentally relevant human cell lines and to detect and interpret epigenomic variation due to a selected set of developmental, physiological, and disease processes.
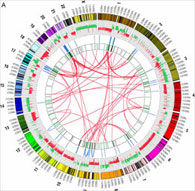
Sites of interchromosomal (red lines) and intrachromosomal (blue lines) rearrangements in an MCF7 breast cancer cell line.
The Genboree Discovery System is the largest software system developed at the BRL. Genboree is a turnkey software system for genomic research. The Epigenomics Data Analysis and Coordination Center and a number of collaborative epigenomic projects use Genboree as the core informatic infrastructure. Other current studies involving Genboree include studies of genome variation in collaboration with HGSC in the context of the NIH 1000 Genomes Project, array CGH studies of structural genome variation, mapping rearrangements in tumor genomes using paired-end tags and sequences, genome analysis.
Our Positional Hashing method, implemented in the Pash program, enables extremely fast and accurate sequence comparison and pattern discovery by employing low-level parallelism. Pash enables fast and sensitive detection of orthologous regions across mammalian genomes, and fast anchoring of hundreds of millions of short sequences produced by massively parallel sequencing technologies. The anchoring is the key step in an increasing diversity of applications of massively parallel sequencing in epigenomics, cancer genomics, and human genome resequencing. For example, by being able to accurately analyze billions of sequence fragments from ChIP-seq, bisulfite-seq and other *-seq assays performed in the course of the NIH Epigenomics Roadmap Initiative project, we anticipate to be able to gain insights into the interplay between human genetic and epigenetic variation.
We are developing comprehensive, rapid, and economical methods for detecting and interpreting recurrent chromosomal aberrations in cancer using massively parallel sequencing technologies. One of the key objectives is to integrate the rearrangements with other types of genomic information gathered by The Cancer Genome Atlas (TCGA) and related projects and to identify functionally significant recurrent rearrangements in cancer that are positively selected during pathological evolution of cancer cells. Recurrent chromosomal aberrations are best understood in leukemias, lymphomas, and sarcomas. However, recent evidence suggests that carcinomas also contain important recurrent rearrangements, which are not detectable using current methods. We anticipate that the study of highly rearranged genomes such as those found in breast, ovarian, and prostate cancer will particularly benefit from the increase in resolution that we aim to achieve.